
Energetic studying for nanoscale construction
Now we have developed a complete dataset of atomistic constructions and quantum-mechanical reference information for the binary Si–O system, in addition to an interatomic potential fitted to that database within the atomic cluster enlargement (ACE) framework33,34,35. We initialised the protocol with two present datasets for silicon (Bartók et al. 30) and silica (Erhard et al. 31) respectively, and we then step by step explored the related configurational area utilizing the active-learning workflow illustrated in Fig. 1. Quantum-mechanical reference (“coaching”) information for energies and forces had been obtained with the strongly constrained and appropriately normed (SCAN)36 alternate–correlation useful for DFT, which reveals good efficiency for elemental silicon37 and the assorted silica polymorphs31.
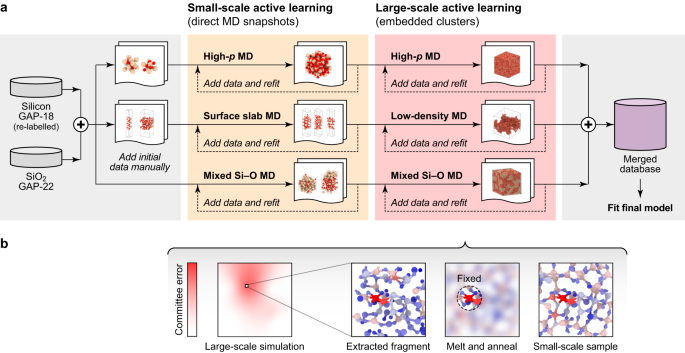
a Overview of the process to acquire the database. After merging constructions from the Si-GAP-18 (ref. 30) and SiO2-GAP-22 (ref. 31) databases, the method was cut up into three tracks, aiming to explain high-pressure (“Excessive-p”) silica, silica surfaces, and combined Si–O techniques with completely different stoichiometric compositions. In every of the tracks, small-scale molecular dynamics (MD) simulations had been used to pattern new constructions by lively learning38. Within the final step, large-scale simulations had been carried out, the place atoms with excessive uncertainty had been recognised by a committee error. b Schematic illustrating the idea of our amorphous matrix embedding strategy. First, we extract the broader atmosphere of an atom with excessive uncertainty (indicated utilizing a colourbar with purple shading: be aware the “Giant-scale simulation” is merely a schematic sketch). Then we preserve the atom of curiosity, in addition to the direct atmosphere mounted, and we soften and anneal the outer atmosphere (indicated by blurring the area exterior the “Mounted” sphere). Because of this, we acquire a small-scale structural mannequin which has the atom of curiosity and its native atmosphere embedded into an amorphous matrix. This pattern could be fed into the coaching database.
Our active-learning workflow follows three foremost tracks: high-pressure bulk silica, silica surfaces, and non-stoichiometric SiOx techniques (Fig. 1a). The person tracks are stored separate throughout preliminary coaching, i.e., they don’t share their newly generated coaching information; nonetheless, ultimately, all constructions are merged into one complete database.
The only subtracks are additional divided into phases. Within the first stage, we added preliminary constructions, e.g., for crystalline high-pressure polymorphs or floor fashions. Within the subsequent stage, we fitted second tensor potential (MTP) models38 to the database and used these MTPs to discover configurational area in MD and to determine new constructions by lively learning39. Energies and forces for brand new constructions had been computed with DFT and added to the database. This course of was iterated till the extrapolation threshold (Supplementary Observe 1B) was not exceeded through the MD trajectories anymore.
The third stage, highlighted in purple in Fig. 1a, is an important a part of our workflow, and is predicated on large-scale simulations in every monitor. We used 2–4 MTPs educated on the identical database to estimate a per-atom committee error, as is usually executed for neural-network potentials40. For atoms with excessive uncertainty (Supplementary Observe 1C), we extracted the environments into smaller, “DFT-sized” cells by an strategy that we name amorphous matrix embedding (Fig. 1b). After figuring out an atom with excessive uncertainty, we lower out a dice containing the corresponding atmosphere of the atom. This dice has a measurement which is possible for performing DFT computations; it’s typically chosen bigger than twice the cut-off of the potential. After extracting the dice, the atoms inside the cut-off radius of the atom with excessive uncertainty are stored mounted. The remaining construction is melted in an ML-MD simulation to create an amorphous matrix and clean boundaries. Particulars of the process could be present in Supplementary Observe 1C. We be aware very current, associated approaches to isolating fragments for lively studying primarily based on minimising the uncertainty for boundary atoms41,42.
The ultimate database was obtained by merging the info of all tracks collectively, together with some extra samples equivalent to clusters and vacancies. This database incorporates 11,428 constructions with a complete of ≈1.3 million atoms (Supplementary Desk 1). For validation, we held out 5% of those constructions from coaching, chosen at random.
Efficiency
The ultimate potential is a posh non-linear ACE mannequin, obtained by summation of 1 linear and 7 non-linear ACE phrases (Strategies). This strategy permits a extra versatile description than only a linear or Finnis–Sinclair-like embedding, at solely reasonably increased computational expense. The ensuing potential has a test-set root imply sq. error (RMSE) of 16.7 meV atom−1 for energies and 306 meV Å−1 for forces. These errors are averaged over the total dataset, nonetheless, and so they don’t seem to be in themselves adequate to characterise the standard of the potential. For instance, they seek advice from a extremely heterogeneous set of constructions, with goal power values spanning greater than 8 eV atom−1, and a spread of forces of 40 eV Å−1 coated by the database. Moreover, the numerical accuracy of the potential in sure elements of configurational area (e.g., crystalline polymorphs) is much extra essential than in others (e.g., liquid and amorphous constructions).
In Desk 1, we subsequently present the efficiency of our mannequin on completely different separate check units. The advanced non-linear ACE is in comparison with our earlier silica GAP mannequin described in ref. 31 (“SiO2-GAP-22” within the following), and in addition to less complicated ACE fashions fitted to the brand new database utilizing linear and Finnis–Sinclair-like embeddings, respectively. Certainly, the advanced non-linear ACE potential is the one one among the many three which achieves comparable errors to SiO2-GAP-22 for amorphous and crystalline silica constructions. In distinction, for amorphous elemental silicon, mixed-stoichiometry in addition to high-pressure phases the advanced non-linear ACE is considerably extra correct than SiO2-GAP-22, since these constructions will not be a part of the GAP database. This desk subsequently signifies the principle problem – and its answer – within the current mannequin in comparison with the earlier GAP: each are extremely correct for crystalline (≈1 meV atom−1) and bulk amorphous (≈5 meV atom−1) SiO2, however our ACE mannequin caters to a a lot wider vary of eventualities exterior of the 1:2 stoichiometric composition.
Determine 2 reveals the part diagram of SiO2 calculated by thermodynamic integration43,44 utilizing the ACE potential in comparison with a CALPHAD part diagram from the literature45. The ACE and CALPHAD predictions agree properly all through, and for the boundary between quartz and coesite we observe virtually quantitative settlement. In distinction, the cristobalite and tridymite phases appear to be over-stabilised. At 0 GPa, the melting level is notably overestimated (about 2400 Okay, in comparison with ≈2000 Okay experimentally45); furthermore, the part stability areas of each phases are extra prolonged than within the reference. As an instance the sensitivity of the evaluation to small errors in predicted energies, we added a fictitious power penalty of 5 meV atom−1 for cristobalite and tridymite (Supplementary Fig. 1a); on this case, the transition traces agree significantly better with the CALPHAD reference than earlier than. Additional numerical assessments confirmed that the tridymite–cristobalite transition line, specifically, is strongly affected by small shifts in power (Supplementary Fig. 1b–f). We thus conclude that the quantitative deviation seen in Fig. 2 is because of the inaccuracy of the underlying alternate–correlation useful, fairly than indicating a shortcoming of the ACE strategy. That is an instance of the extra common drawback that any points with the ground-truth methodology (e.g., numerical instabilities) will translate into the ML mannequin.
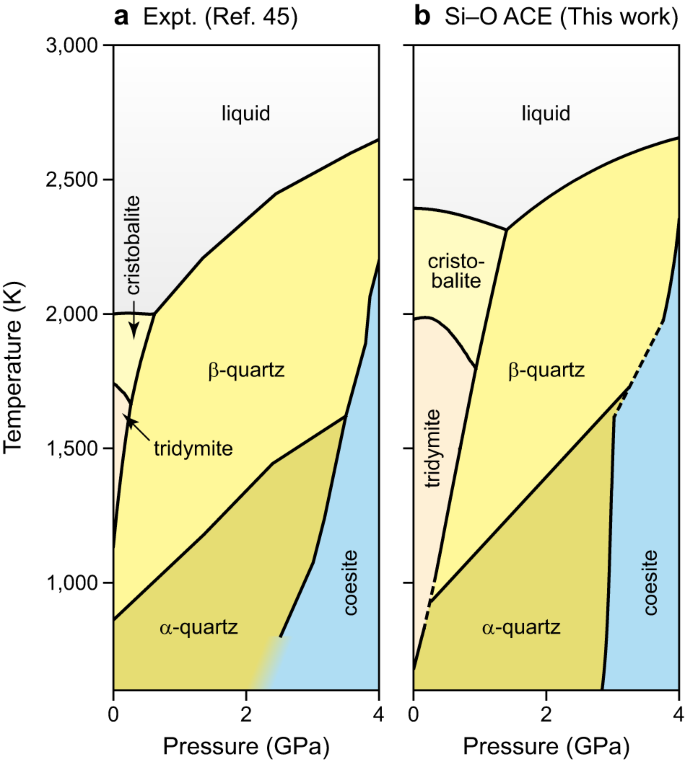
a Part diagram calculated primarily based on experimental information, tailored from the literature (ref. 45). b The identical part diagram calculated primarily based on predictions from our Si–O atomic cluster enlargement (ACE) mannequin and thermodynamic integration. The background shading in several colors emphasises the steadiness areas of various phases. Supply information are supplied as a Supply Information file.
Excessive-pressure structural transitions of SiO2
Determine 3 characterises high-pressure properties of silica. In Fig. 3a, we present power–quantity curves of α-quartz, coesite, stishovite, α-PbO2-type, and pyrite-type silica as predicted by our ACE mannequin and in contrast with DFT information, with which they agree properly. As well as, we examined the behaviour of the mannequin for rosiaite-type silica, which was lately noticed in experiment46 and predicted theoretically47 for direct compression of α-quartz. In distinction to the constructions talked about earlier than, this explicit polymorph shouldn’t be a part of the coaching database. Nonetheless, the ACE mannequin reproduces DFT information for this construction equally properly as for the opposite polymorphs.
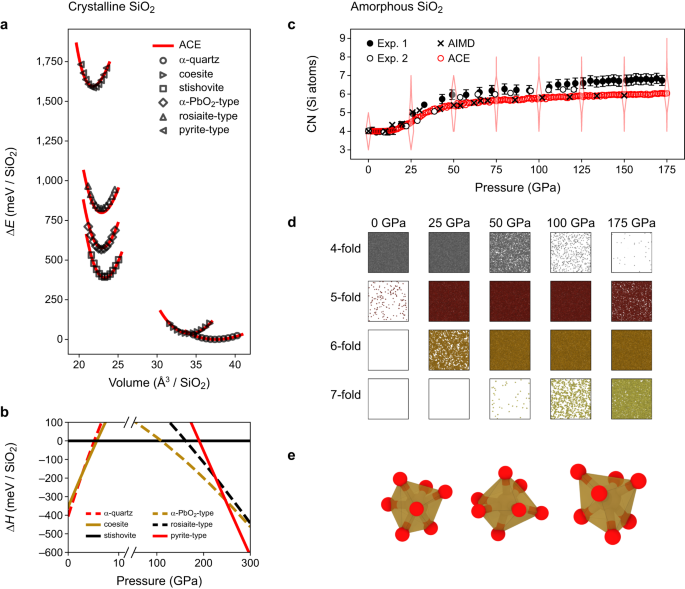
a Power–quantity curves for high-p silica polymorphs with energies referenced to α-quartz (ΔE). Markers point out outcomes from density-functional concept computations with the strongly constrained and appropriately normed alternate–correlation useful (SCAN DFT); traces point out the atomic cluster enlargement (ACE) predictions. b Enthalpy variations (ΔH) for varied silica polymorphs referenced to stishovite. c Compression of a vitreous silica construction. Outcomes for the typical silicon coordination quantity (CN) are in comparison with experimental measurements from ref. 5 (“Exp. 1”) and ref. 50 (“Exp. 2”) and ab initio MD (AIMD) simulations from ref. 51. The distribution of coordination numbers at chosen pressures is indicated by violin plots. d Snapshots of the compression simulation displaying coordination polyhedra for various coordination numbers (solely). e Visualisation of the coordination environments of chosen 7-fold coordinated silicon atoms. Supply information are supplied as a Supply Information file.
Determine 3b reveals an enthalpy–strain diagram at 0 Okay. For decrease pressures, there’s a transition from α-quartz to coesite between 2.5 and three.0 GPa, in line with the expected part diagram (Fig. 2), adopted by a transition to stishovite at 5.5–6.0 GPa. At increased pressures of ≈110 GPa, we observe the transition from stishovite to α-PbO2-type silica. Experimentally, fairly than stishovite (rutile sort), the structurally carefully associated CaCl2 (distorted rutile) sort polymorph of silica is secure. The transition from CaCl2- to α-PbO2-type silica was noticed at 120 GPa and 2400 K48. Provided that our enthalpy information correspond to a temperature of 0 Okay, each values agree properly with one another. For the transition of α-PbO2- to pyrite-type silica, our ACE mannequin predicts a strain of ≈246 GPa, in good settlement with the experimentally decided transition strain of ≈260 GPa at 1800 K49. Lastly, rosiaite-type silica46 is appropriately recognized as metastable over the strain vary studied.
Determine 3c reveals the strain evolution of the typical coordination quantity (CN) of silicon atoms in amorphous silica, extracted from an MD simulation at room temperature and beneath isostatic strain. The ACE outcomes agree properly with experiment as much as about 50 GPa5,50, and with ab initio MD51 outcomes over the entire strain vary. The nice settlement with experiment is especially pronounced for the info from ref. 50. Above 50 GPa, our mannequin underestimates the typical CN: at 175 GPa the experimental estimate is about 7; the ACE simulation predicts it to be 6. Importantly, this doesn’t imply that there aren’t any 7-fold coordinated environments, however there stay some 5-fold coordinated atoms as properly, reducing the typical (Fig. 3d). A potential motive for the great settlement with the ab initio outcome, however the deviation from experiment, could be the restricted time scales in our simulations, which hinder a whole transition into higher-coordinated environments. Furthermore, we be aware that computed X-ray-Raman spectra of the ab initio constructions from ref. 51 are in good settlement with experiment indicating a decrease CN. Different MD simulations additionally confirmed barely decrease CNs than the experimental values52. Determine 3e reveals three completely different 7-fold coordinated environments extracted from the simulations.
A CN of seven in amorphous silica could be shocking, since silicon is sixfold-coordinated in all crystalline silica polymorphs which are secure on this strain vary. Nevertheless, the pyrite-type part, which turns into thermodynamically secure at ≈240–260 GPa, incorporates silicon atoms with a 6+2-fold atmosphere. A current examine discovered sure, however restricted, similarities between these 7-fold environments in glassy and pyrite-type silica52.
In Supplementary Fig. 2, we present two extra structural fingerprints which have been generally analysed in experiment: the place of the primary sharp diffraction peak and the Si–O bond size. For each circumstances, our simulations present good settlement with experiment.
SiO2 surfaces and aerogels
Determine 4 assessments the power of the potential to precisely predict floor energies. We start with validation for various α-quartz surfaces: we created floor slab fashions, relaxed them with the ACE potential, and evaluated the energetics, and subsequently the floor power per space, with DFT single-point computations (Fig. 4a). The ACE outcomes agree properly with DFT, particularly contemplating that the coaching database doesn’t comprise all of the floor terminations proven. The ACE mannequin can also be in a position to predict the steadiness of the reconstructed α-quartz (001) floor (Supplementary Fig. 3a, b). While these floor energies could be computed with DFT, real looking amorphous floor energies are rather more tough to acquire, because of the required system sizes. Due to this fact, Fig. 4b validates the potential on 125 small-scale floor constructions of amorphous SiO2, every containing 192 atoms. The floor fashions are created primarily based on bulk constructions from ref. 31; the latter had been generated in soften–quench simulations with completely different interatomic potentials and subsequently span a spread of energies. Whatever the beginning construction, the ACE mannequin captures the floor power for all slab fashions very properly: the whole RMSE is about 0.01 eV/Å2, and solely a slight underestimation in comparison with DFT is seen. Furthermore, there aren’t any clear outliers though the assorted floor energies point out a big variety of the floor constructions.
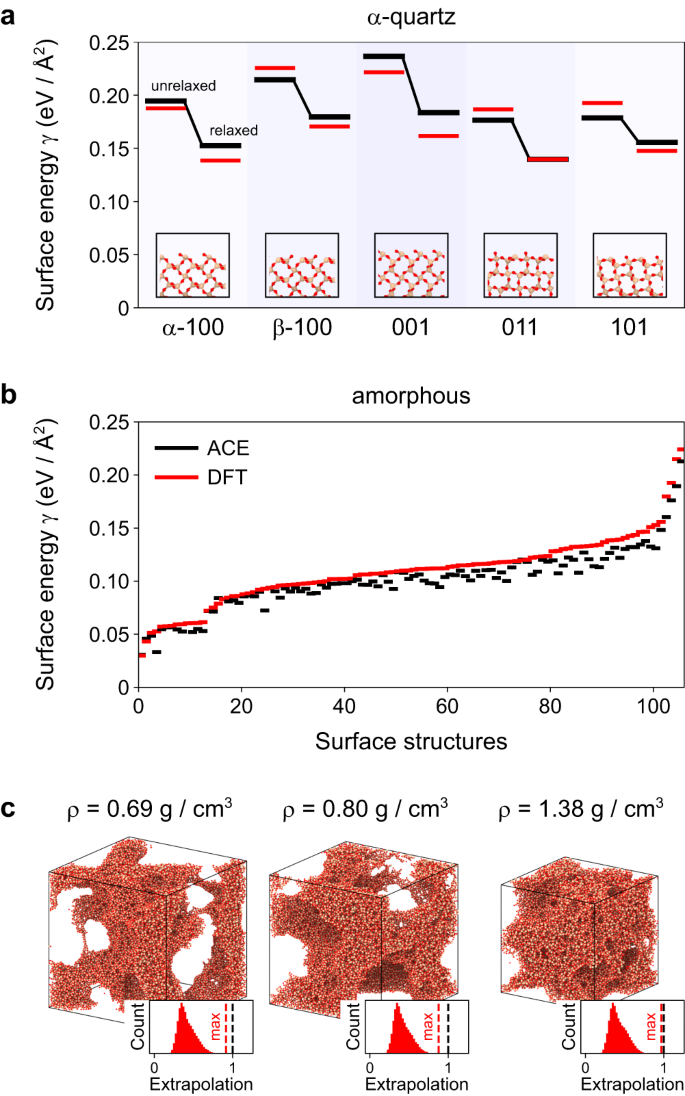
a Floor energies γ of assorted α-quartz surfaces in relaxed and unrelaxed state. The black traces present the atomic cluster enlargement (ACE) outcomes whereas the purple traces present the density useful concept (DFT) outcomes for the unrelaxed constructions and DFT re-relaxed constructions. The construction photos present the unrelaxed surfaces. b Floor energies of amorphous fashions calculated with ACE and DFT. Amorphous constructions had been taken from ref. 31, and have been created utilizing varied interatomic potentials: the BKS23, CHIK25, SiO2-GAP-2231, Munetoh59, and Vashishta24 fashions. The constructions had been relaxed with ACE and sliced at varied factors, adopted by one other ACE rest. DFT floor energies had been decided with out additional rest. c Exemplary porous amorphous silica constructions with varied densities ρ. Moreover, we present the distribution of the linear extrapolation grade41 and the utmost extrapolation grade (purple line) for every construction. For all constructions, the utmost extrapolation grade is under one. Supply information are supplied as a Supply Information file.
The amorphous surfaces proven are already very advanced, however in actuality they’re usually not flat as right here. They’ve curvature, for instance when occurring inside pores, and such advanced constructions can now not be instantly validated with DFT. Determine 4c subsequently reveals how properly atomic environments in varied porous amorphous constructions are coated by the dataset. These constructions had been ready by straining amorphous constructions at elevated temperatures to the specified density. To validate the efficiency of the potential on this mannequin, we present the linear extrapolation grade in response to the maxvol selection41,53. An extrapolation grade above 1 corresponds to atomic environments that haven’t been coated within the coaching database. This doesn’t imply that the potential is now not dependable, as there’s a sure vary of kind of dependable extrapolation, however because the extrapolation grade will increase, non-physical behaviour and failure of the potential turns into an increasing number of likely39. For all porous constructions, no matter density, we discover that the utmost extrapolation worth is lower than 1. Thus, we observe no pronounced extrapolation in any of the circumstances thought of, indicating an correct description of the potential for a wide range of curved surfaces. In Supplementary Fig. 3e, f, we reveal the effectiveness of the ACE potential in reconstructing manmade amorphous surfaces. To this finish, we lower out a spherical cavity from an amorphous bulk construction. Upon heating, the quantity of incorrectly organized atoms close to the floor (<3 Å) decreases over time, indicating that the floor atoms are present process vital reconstructions.
Elemental silicon
While our ACE mannequin is designed for the binary Si–O system, we present in Desk 2 the efficiency for diamond-like elemental silicon in comparison with each DFT and experiment. The majority modulus may be very properly reproduced, whereas the emptiness formation power is underestimated by about 30%. The experimental floor energies are properly recovered by the ACE mannequin, however this can be partly attributable to serendipity, as a result of the SCAN ground-truth information present poorer high quality (Desk 2). Furthermore, the potential captures the reconstruction of the diamond-type silicon (100) floor (Supplementary Fig. 3c, d). We additionally computed the linear thermal enlargement coefficient of diamond-type silicon within the quasiharmonic approximation, discovering virtually excellent settlement between the ACE prediction and experiment; specifically, the weird destructive enlargement coefficient under 130 Okay is reproduced (Supplementary Fig. 4). Lastly, soften–quench simulations had been carried out to generate a-Si constructions (Supplementary Fig. 5). The settlement with the experimental construction issue is pretty much as good as that for a GAP-18-generated construction from ref. 54. As well as, we’re in a position to obtain decrease quenching charges with the ACE than with the GAP, and for quenching charges as little as 1010 Okay s−1, we noticed crystallisation.
In comparison with Si-GAP-1830, we observe increased errors with respect to the reference information (Desk 2). This isn’t a principal shortcoming of ACE in comparison with GAP: it was already proven that it’s potential to suit an ACE potential with comparable numerical accuracy as Si-GAP-18 to the identical coaching database34. As an alternative, the decrease accuracy could be brought on by the extension of the database to a second component, in comparison with the Si-GAP-18 one, and its sturdy concentrate on the SiO2 a part of the configurational area. We assume that this causes, in flip, a much less correct description of the configurational area of the basic species. Bigger coaching databases would possibly assist to beat this problem sooner or later. As a result of decrease accuracy in reproducing the SCAN information, our potential has some shortcomings for higher-pressure constructions: the bc8 part is erroneously predicted to be secure at elevated strain (Supplementary Fig. 6), and upon compressing a-Si we don’t observe the eventual crystallisation that’s described by Si-GAP-18 (Supplementary Fig. 7)55. We emphasise that very-high-pressure silicon phases weren’t the scope of the current work – as a substitute, we concentrate on the correct description of ambient-pressure silicon as a constituent a part of combined binary phases and nanostructures.
SiO and combined silicon–silica techniques
While the outcomes to date have served to reveal the usefulness of the strategy – each when it comes to growth of datasets and the becoming inside the ACE framework – we at the moment are in a position to examine an precise utility drawback. To this finish, Fig. 5a reveals structural fashions of SiO. Experimentally, amorphous SiO is obtained by deposition of SiO from the fuel phase56. In distinction, we created our fashions by soften–quench simulations. SiO phases are identified to be metastable with respect to Si and SiO2. For instance, a current DFT-based crystal-structure prediction examine explored potential ordered phases of homogeneous SiO, and located that these are metastable in comparison with a mix of crystalline Si and SiO257. We verified that our ACE potential equally reproduces the metastability of the ambient-pressure phases (Supplementary Fig. 9), and that it precisely predicts related Si–SiO2 interface energies (Supplementary Fig. 10). In good settlement with these outcomes, our melt-quenched constructions present a transparent segregation between a-Si-like (blue) and a-SiO2-like areas (purple). With lowering quench price, the variety of silicon grains decreases whereas their measurement will increase. Determine 5b reveals the construction issue, S(q), for the construction quenched with 5 × 1012 Okay s−1 (information for the opposite constructions are proven in Supplementary Fig. 11), which agrees finest with the experimental information from ref. 15. Determine 5c reveals the ratio between the quantity of the silicon grains divided by the interface space. Within the approximation of spherical particles, the grain diameter is d = 6 ⋅ VSi,grains/Ainterface. From this we will estimate common grain diameters between 24 and 54 Å for the examined quench charges. These grain diameters agree very properly with transmission electron microscopy measurements, which indicated diameters of 30–40 Å14.
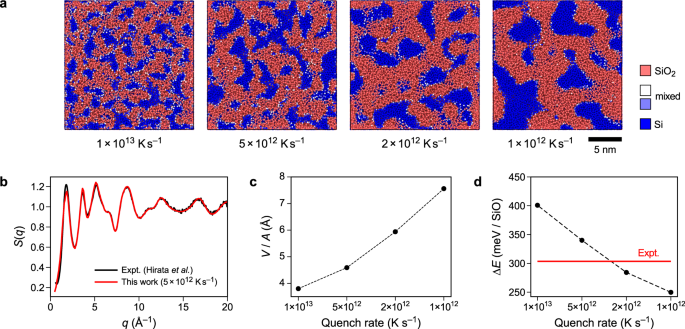
a Visualisation of SiO constructions generated by quenching from the soften at charges between 1013 and 1012 Okay s−1. Color-coding is predicated on the nearest-neighbour depend as much as 2.0 Å, taken to correspond to the Si–O coordination numbers, that are 4 in SiO2 and 0 in elemental silicon. Accordingly, SiO2-like and Si-like areas are indicated in purple and blue, respectively. b Construction issue, S(q), as a perform of the wave vector, q, for the 5 × 1012 Okay s−1 simulation, decided at 300 Okay. c Relation between grain quantity, V, of the silicon grains and the interface space, A, between silicon and silica. With growing quench price, the grain measurement of the constructions decreases. d Power of the SiO constructions referenced to α-quartz and to diamond-type silicon (ΔE), in comparison with the experimental customary enthalpy of formation for SiO58. The predictions proven are sturdy with respect to system measurement (Supplementary Fig. 8). Supply information are supplied as a Supply Information file.
Determine 5d reveals the surplus energies of the constructions referenced to to α-quartz and diamond-type silicon. The SiO constructions had been relaxed by optimisation of the cell measurement in addition to the atomic positions at 0 Okay. As experimental reference, we present the usual enthalpy of formation of SiO58. The constructions generated utilizing quench charges of 5 × 1012 and a pair of × 1012 Okay s−1 have energies akin to experiment. Certainly, we will even create constructions which are energetically extra beneficial than in experiment, noting once more that our process to supply the constructions deviates considerably from the experimental one.
However is that this actually an enchancment in comparison with present, empirically fitted interatomic potentials? We examined the Munetoh potential59 and a charge-optimised many-body (COMB) potential29 for a similar process to generate structural fashions of SiO. The Munetoh potential yielded a homogeneous construction with out observable segregation into silicon and SiO2, and the ensuing construction issue (Supplementary Fig. 11c) deviates strongly from experiment. For the COMB potential, we noticed pore formation at elevated temperatures, lastly leading to a strongly elevated simulation-cell measurement. Due to this fact, we solely equilibrated our best-matching construction at room temperature and analysed the change in construction issue (Supplementary Fig. 11f): once more, we noticed a robust deviation from experiment, indicating that the construction may be very completely different from the ACE mannequin prediction.
Crystallisation of silicon in amorphous SiO
Silicon in silicon monoxide is experimentally identified to crystallise above 850 °C60. Determine 6 illustrates simulations of such crystallisation processes. The SiO constructions proven in Fig. 5 had been heated to 1400 Okay, inflicting the silicon-rich areas to soften whereas the silica matrix remained stable. The constructions had been then quenched to 1200 Okay inside 20 ns (Fig. 6a). By way of this cooling course of, we seen crystallisation within the silicon-rich areas of the SiO construction. Particulars are proven in Supplementary Fig. 12, indicating how crystallisation begins from two seeds, showing shortly after one another and propagating all through the construction. Earlier than thermal therapy, all constructions present practically no signal of crystallinity, whereas afterwards, the constructions with bigger silicon grains do (Fig. 6c–f). This additionally impacts the construction issue: these techniques with no or solely a small quantity of crystallinity (quench charges: 1 × 1013 and 5 × 1012 Okay s−1) present construction elements which are nonetheless much like the experimental construction issue of SiO (Fig. 6g), whereas these with bigger quantities of crystalline silicon (1 × 1012 and a pair of × 1012 Okay s−1) present distinct S(q) peaks that point out crystallinity (Fig. 6h). This comparability permits us to exclude the incidence of enormous quantities of crystalline silicon in silicon monoxide samples, given the experimental S(q) (cf. Fig. 6g and ref. 15).
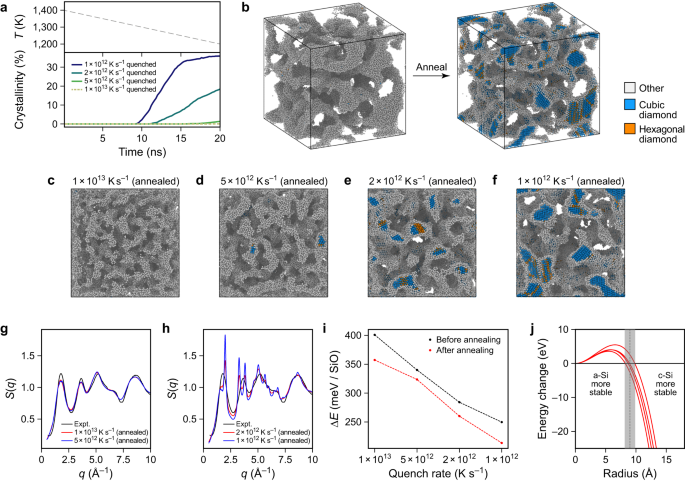
a Evolution of crystallinity over time for the thermal therapy of the completely different SiO constructions. The temperature (T) change is identical in all circumstances, and characterised within the higher panel. b The silicon community inside the SiO construction quenched at 1012 Okay s−1: for this picture, we solely present Si atoms which aren’t bonded to a couple of O atom (2 Å cut-off). The color coding signifies the kind of construction recognised by polyhedral template matching (root imply sq. deviation cut-off: 0.1). The preliminary construction is totally amorphous; after thermal therapy, crystalline areas are noticed. c–f All constructions from Fig. 5a after thermal therapy. g, h X-ray construction elements S(q) as perform of the wave vector q for the constructions proven in panels c–f. For the constructions with excessive crystallinity, the construction issue incorporates extra distinct peaks. i Computed power of formation ΔE for constructions earlier than and after warmth therapy. j Power change of a spherical silicon inclusion inside an amorphous silica matrix by crystallisation. We used interface energies from manually constructed interface constructions generated utilizing amorphous fashions from 4 completely different quench charges. Due to this fact, we present 4 separate information sequence as traces. Supply information are supplied as a Supply Information file.
Determine 6i reveals that each one constructions are energetically extra beneficial after the thermal therapy. Nevertheless, despite the fact that one would possibly anticipate that the stronger crystallised constructions acquire extra power, we observe no direct connection between the power acquire and the extent of crystallinity after warmth therapy. The reason being doubtless that for the fast-quenched construction (1 × 1013 Okay s−1), the silicon-rich areas could be not completely organized. Thermal therapy thus lowers the interface power and the inner power of the amorphous silicon area. For the crystallised constructions there may be, on one hand, an power acquire attributable to crystallisation however, alternatively, an power loss because of the increased interface power between crystalline silicon and the silica matrix in comparison with that of a-Si and the silica matrix (cf. Supplementary Fig. 13). Based mostly on these interface energies, we constructed a easy physically-based mannequin (Strategies) to calculate the power acquire of a spherical silicon inclusion in an amorphous silica matrix by crystallisation. This power acquire is proven in Fig. 6j for interface energies taken from 4 pairs of manually constructed interfaces. The distinction between the fashions is fairly small, indicating that the minimal radius of a spherical inclusion needs to be round 9 Å to be energetically beneficial in a crystalline state. We be aware that the manually constructed fashions don’t have the identical interface orientations as within the SiO constructions and are additionally not completely relaxed, inflicting an overestimation of the interface energies – as seen, for instance, within the distinction between the interface energies of the SiO constructions and of the manually constructed a-Si–a-SiO2 interface in Supplementary Fig. 13d. Nevertheless, since for our mannequin the distinction between the interface power between a-Si–a-SiO2 and c-Si–a-SiO2 is the one related amount, we assume that these results partially cancel.





{kind=link}